Hier dreht sich alles um den
Alpha-1-Antitrypsin-Mangel
(AATM) bei Kindern!




Unser Team der Unikinderklinik Bonn

„Herzlich Willkommen auf der Internetseite des Alpha-1-Kindercenters der Universitätsklinik Bonn.“
Ihr Rainer Ganschow & Team
Lernen Sie unser Team kennen und erfahren Sie mehr über unsere Motivation, unsere Tätigkeiten und Ziele. Informieren Sie sich über die Erkrankung und über das Alpha-1-Register für Kinder und Jugendliche, welches seit Anfang 2024 als App genutzt werden kann.






Motivation
Der Alpha-1-Antitrypsin-Mangel im Kindesalter liegt unserem Team ganz besonders am Herzen. Nach wie vor werden viele Kinder und Jugendliche zu spät diagnostiziert. Dies möchten wir ändern, um möglichst allen Kindern eine zeitnahe und professionelle Betreuung zu ermöglichen.
Auch wenn viele betroffene Patienten mit Alpha-1-Antitrypsin-Mangel einen relativ guten klinischen Verlauf haben, so sehen wir leider ebenfalls schwere Verläufe. Aber auch bei schwerem Verlauf können wir effektiv helfen, vor allem wenn die Diagnose rechtzeitig gestellt wird.

Tätigkeiten & Ziele
In den letzten Jahren haben wir in Bonn eine Spezialambulanz für Kinder und Jugendliche mit Alpha-1-Antitrpysin-Mangel aufgebaut und betreuen hier mittlerweile knapp 50 Patienten mit schwerem Mangel so wie viele Weitere mit weniger starkem Mangel. Eine dauerhafte Betreuung der Patienten und ihrer Familien bis zum Übergang ins Erwachsenenalter ist uns ein großes Anliegen. So sind eine bestmögliche fachliche Betreuung, Aufklärung der Familien und strukturierte Transition (Übergabe eines Patienten an Erwachsenenmediziner) erreichbar.
Neben der klinisch-medizinischen Betreuung unserer Alpha-1-Patienten haben wir es uns zum Ziel gesetzt Forschungsprojekte zu initiieren oder zu unterstützen, die in der Zukunft zu einer noch besseren Versorgung von Kindern und Jugendlichen mit Alpha-1-Antitrpysin-Mangel führen sollen. Hierfür arbeiten wir mit mehreren Arbeitsgruppen aus anderen deutschen Kliniken zusammen.
2021 wurde Herr Dr. Katzer für eine gemeinsame Übersichtsarbeit mit den Aachener Kollegen Prof. Strnad und PD Dr. Hamesh zum Thema Alpha-1-Antitrpysin-Mangel mit dem Paul Caspar Tyrell Preis ausgezeichnet. Dieser wird vom Verein leberkrankes Kind e.V. gestiftet.
Ein Herzensprojekt ist zudem das deutsche Register für Kindern und Jugendlichen mit Alpha-1-Antitrpysin-Mangel. Nach der Entwicklung einer Register-App, konnte das neu aufgelegte Alpha-1-KIDS Register Anfang 2024 online gehen. Mit dem App-basierten Register wollen wir den Familien die Meldung möglichst unkompliziert und ohne viel Aufwand ermöglichen. Wir sind überzeugt davon, dass wir mit dem Register viele wichtige Informationen erhalten können, die uns dabei helfen werden, die Versorgung unserer Alpha-1-Patienten zu verbessern. Jeder gemeldete Patient hilft! Im März 2024 wurde die innovative Register-App bereits mit dem Alpha1 Science Award der Firma CSL Behring prämiert.
Veröffentlichungen unserer Arbeitsgruppe
Update Alpha-1-Antitrypsin-Mangel
D. Katzer, A. Briem-Richter, A. Weigert, E. Lainka, S. Dammann, E. D. Pfister, S. Wirth, R. Kardorff, R. Ganschow
Pi*ZZ-related liver disease in children and adults—narrative review of the typical presentation and management of alpha-1 antitrypsin deficiency
D. Katzer, R. Ganschow, P. Strnad, K. Hamesch
Alpha-1-Antitrypsin Deficiency in Children—Unmet Needs Concerning the Liver Manifestation
J.Lemke, A. Weigert, S. Bagci, M. Born, R. Ganschow, D. Katzer
Unklare Transaminasenerhöhung – schon an Alpha-1-Antitrypsin Mangel gedacht?
A. Weigert, R. Ganschow, D. Katzer
Pädiatrie-Podcast Consilium: Alpha-1-Antitrypsin-Mangel – AATM (#46)
R. Ganschow

Transition
Mit dem Erwachsenwerden wird die sogenannte Transition, die Übergabe an Ärzte fur Volljährige Patienten, notwendig. Hierbei gilt es zu beachten, dass neben der Leber auch der Lunge eine zunehmend größere Bedeutung zukommt. Auf der folgenden Homepage finden Sie alle Alpha-1-Center für Erwachsene in Deutschland: Link
Spezialambulanz
Sie möchten einen Termin in unserer Spezialambulanz für Kinder und Jugendliche mit Alpha-1-Antitrypsin-Mangel vereinbaren oder haben eine Frage?



Allgemeine Informationen zum Alpha-1-Antitrypsin-Mangel
Der Alpha-1-Antitrypsin-Mangel, kurz AATM, ist eine erbliche, d.h. genetische Erkrankung. Damit sind Sie oder Ihr Kind aber nicht allein: die Wahrscheinlichkeit mit einem schweren AATM geboren zu werden liegt in Europa bei bis zu 1:2000. Obwohl ein Alpha-1-Antitrypsin-Mangel heutzutage leicht diagnostiziert werden kann (eine Blutentnahme genügt), wird die Diagnose lediglich bei 10-15% der Betroffenen im Laufe ihres Lebens gestellt.
Bei gesunden Menschen wird das Protein Alpha-1-Antitrypsin in der Leber gebildet und gelangt von dort ins Blut. Wenn ein Alpha-1-Antitrypsin-Gendefekt vorliegt, bildet die Leber ein fehlerhaftes Alpha-1-Antitrypsin.
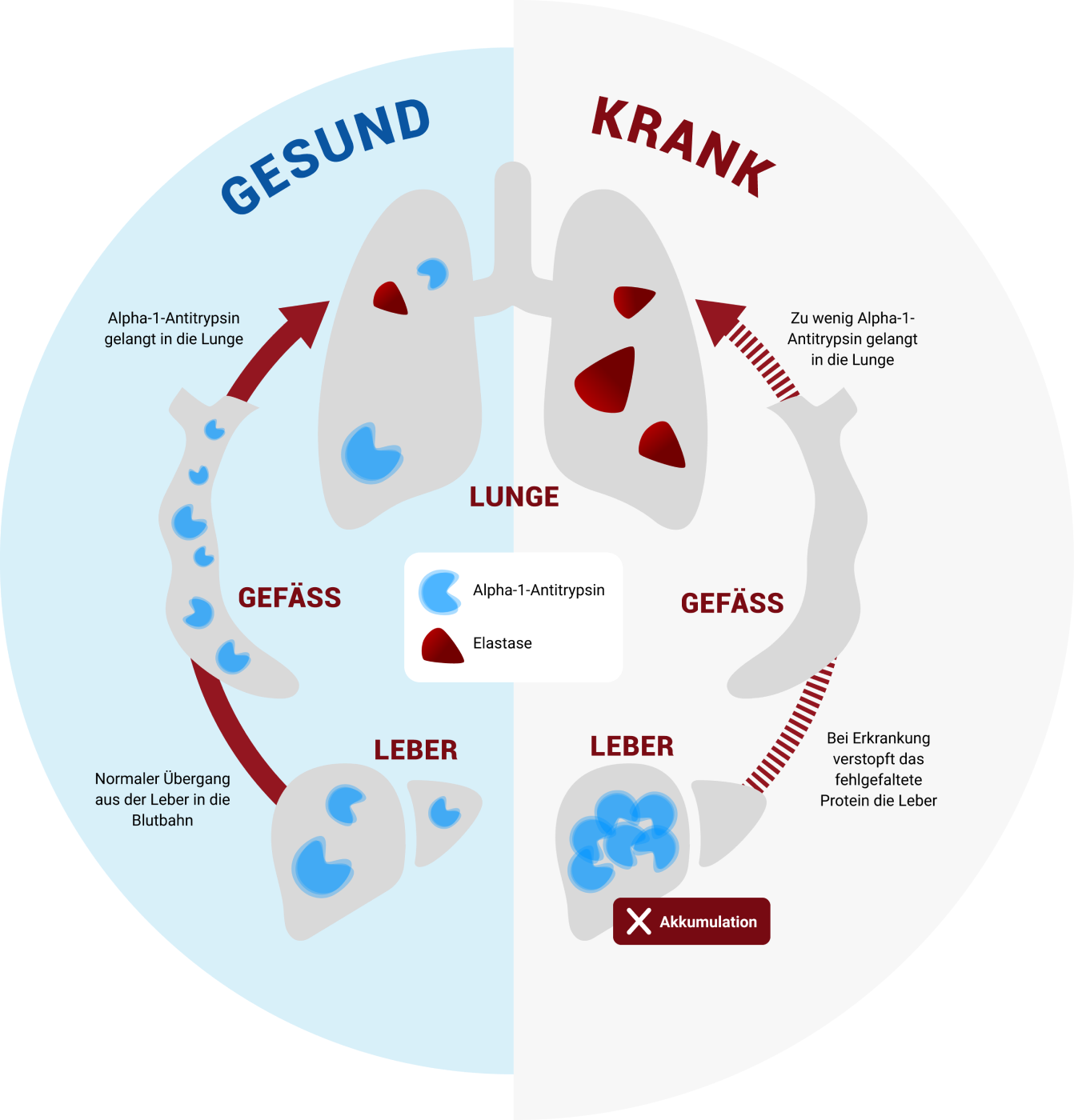
Das kann zu zwei Problemen führen:
 In der Leber:
In der Leber:Das fehlerhafte Protein verbleibt in der Leber und führt dort zu einer Art Verstopfung (Akkumulation). Dadurch kann es zu einer Schädigung der Leberzellen kommen, welche die Leber in ihrer Funktion einschränken kann. Dieses Problem kann bereits bei Neugeborenen, Säuglingen und Kleinkindern oder auch erst später auftreten.
 In der Lunge:
In der Lunge:Des Weiteren gelangt das Protein durch die Akkumulation in der Leber nicht an sein eigentliches Ziel: die Lunge. Diese ist zahlreichen Außeneinflüssen ausgesetzt. Durch Viren und Bakterien, aber z.B. auch durch Tabakrauch und Feinstaub, wird sie gereizt. Hier kommen sogenannte Elastasen als Teil der normalen Abwehrreaktion ins Spiel. Diese Elastasen reagieren unspezifisch, d.h. sie unterscheiden nicht zwischen körperfremd und körpereigen.
Bei gesunden Menschen reguliert das Alpha-1-Antitrypsin die Elastasen und kann eine überschießende Reaktion dieser verhindern. Menschen mit einem Alpha-1-Antitrypsin-Mangel hingegen haben diesen Schutz nicht oder nur unzureichend, wodurch die Lunge im Laufe des Lebens geschädigt wird. Erste lungenspezifische Symptome treten zumeist aber erst ab dem mittleren Erwachsenenalter auf.
Links zu wichtigen Vereinen
Vererbung

Gene enthalten den Bauplan für ein Protein (Eiweißmolekül) und liegen im menschlichen Erbgut in zwei Kopien (zwei Allelen) vor – so auch das Gen für das Alpha-1-Antitrypsin. Je ein Allel stammt dabei vom Vater und eins von der Mutter.
Das normale Allel für Alpha-1-Antitrypsin wird als „M“ und die am häufigsten vorkommende Mutation als „Z“ bezeichnet. In Deutschland trägt ca. jeder Fünfzigste eine solche Veränderung im AATM-Gen in sich und kann sein/ihr Z-Allel weitervererben. Heterozygote Träger („MZ“, also mit einem gesunden und einem veränderten Allel) haben meist keine Beschwerden und wissen in der Regel nichts von ihrer Trägerschaft. Sind beide Elternteile Träger des Z-Allels, besteht die Möglichkeit, dass das Kind zwei Z-Allele vererbt bekommt. Dies führt zu einem schweren, sogenannten homozygoten, Alpha-1-Antitrypsin-Mangel (ZZ).
Flyer und Broschüren





So finden Sie uns
Eltern-Kind-Zentrum (ELKI), Gebäude 30
Venusberg-Campus 1
53127 Bonn
Google Maps
Linie 600: Haltestelle Uniklinikum Nord
Linie 601: Haltestelle Uniklinikum Hauptpforte
Parkplätze stehen im Parkhaus Mitte direkt am Eingang des UKB-Geländes zur Verfügung.
Nutzen Sie unsere Navi-App für Ihr Mobiltelefon, um sich schnell auf dem Gelände des Universitätsklinikums zurechtzufinden. Dazu scannen Sie bitte einfach den untenstehenden QR-Code ein und laden sich die Navi-App herunter.

Universitätsklinikum Bonn
Eltern-Kind-Zentrum (ELKI), Gebäude 30
Allgemeine Pädiatrie
Venusberg-Campus 1
53127 Bonn
Prof. Dr. Rainer Ganschow
Telefon: +49 (0) 228 287 33213
E-Mail: alpha1kinderzentrum@ukbonn.de